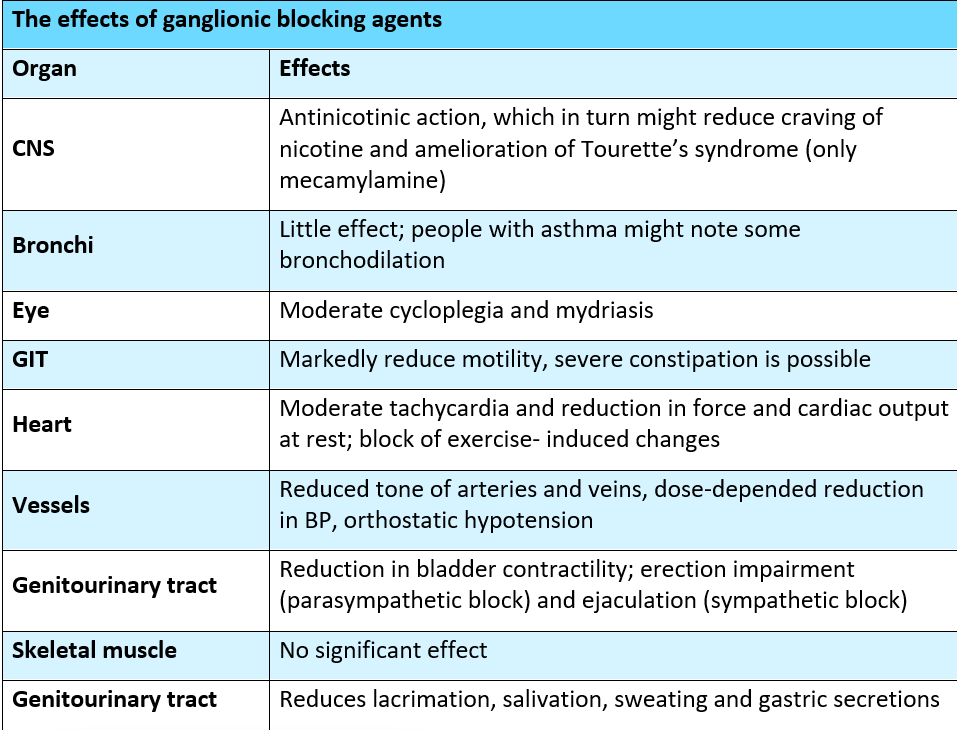
1. What do we mean by GANGLIONIC BLOCKERS?!
2. What do we mean by NEUROMUSCULAR-BLOCKING AGENTS?!
3. Nondepolarizing (competitive) blockers
In the Cholinergic Antagonists article, we mentioned that in addition to antimuscarinic agents there are also ganglionic blockers and neuromuscular-blocking agents which also antagonize the receptors, (muscarinic and nicotinic) subgroups on the basis of their specific receptor affinities. In this article, we will focus on these two remaining cholinergic antagonist agents.
In both sympathetic and parasympathetic ganglia, acetylcholine is the primary excitatory neurotransmitter. Drugs that block acetylcholine synthesis (such as hemicholinium) or acetylcholine release (botulinum toxin, procaine) can disrupt ganglionic transmission, but drugs that act on cholinergic receptors in the ganglia are more selective. Therefore:
Ganglion-blocking agents are competitive antagonists of the action of acetylcholine and similar agonists, specifically acting on the nicotinic receptors of both the parasympathetic and sympathetic autonomic ganglia. Some members of the group also block the nicotinic cholinoceptor-gated ion channel. Because they can block all autonomic outflow, ganglion-blocking drugs are important and used in pharmacologic and physiologic research. The other drugs mentioned in this category, with the exception of nicotine, are nondepolarizing, competitive antagonists. Nondepolarizing blockers' responses are complex and most often unpredictable. Because of their lack of selectivity, ganglionic blockade is rarely used therapeutically, but is frequently used as a tool in experimental pharmacology.
Blockers of ganglionic nicotinic receptors function as competitive pharmacologic antagonists, though there is evidence that some may also block the pore of the nicotinic channel itself. These medications were the first to be approved for the treatment of hypertension.
For this disease, hexamethonium (C6, a prototype), mecamylamine, and several other ganglion blockers were widely used. Unfortunately, the side effects of ganglion blockade in hypertension are so severe (both sympathetic and parasympathetic divisions are blocked) that patients could not tolerate them for long periods of time. Trimethaphan was the most recently used ganglion blocker in clinical practice, but this is also on the verge of being phased out. It is lipid-insoluble, inactive when taken orally, and has a short half-life. It was administered intravenously to treat severe accelerated hypertension (malignant hypertension) as well as to produce controlled hypotension. These drugs are still used in studies.
Recently, there has been a lot of focus on nicotinic receptors in the CNS and their relationship to nicotine addiction and Tourette's syndrome. Contrary to popular belief, nicotine (in the form of nicotine gum or patches), varenicline (a partial agonist administered orally), and mecamylamine, a nicotinic ganglion blocker that enters the CNS, have all been shown to aid in smoking cessation.
Ganglion blockers cause significant venous pooling because they disrupt sympathetic control of venous tone; postural hypotension is a major manifestation of this effect. Dry mouth, blurred vision, constipation, and severe sexual dysfunction are among the other side effects of ganglion-blocking drugs. As a result, ganglion blockers are used infrequently.
Nicotine is the main alkaloid found in tobacco (Nicotiana tabacum); it acts as an agonist on both the NN and NM subtypes of nicotinic cholinergic receptors (NRs). Depending on the dose, nicotine depolarizes autonomic ganglia, causing stimulation and then paralysis of all ganglia. The stimulatory effects are complex, resulting from increased neurotransmitter release as a result of effects on both sympathetic and parasympathetic ganglia. Increased dopamine and norepinephrine release, for example, maybe associated with both pleasure and appetite suppression. The overall response of a physiologic system is a sum of nicotine's stimulatory and inhibitory effects. These include increased blood pressure and heart rate (due to transmitter release from adrenergic terminals and the adrenal medulla), as well as increased peristalsis and secretions. Because of ganglionic blockade, blood pressure falls at higher doses, and activity in the GI tract and bladder musculature ceases.
Nicotine is a poison with a variety of negative effects that is found in cigarette smoke. It is significant in the context of smoking and tobacco chewing; its only clinical indication is for short-term nicotine replacement in tobacco-free subjects. Ganglionic stimulants have no therapeutic application because no useful purpose can be served by stimulating both sympathetic and parasympathetic ganglia at the same time, so it has no therapeutic benefit and is harmful to health.
These medications inhibit cholinergic transmission between motor nerve endings and nicotinic receptors on skeletal muscle. They have some chemical similarities to acetylcholine and act as antagonists (nondepolarizing type) or agonists (depolarizing type) at the receptors on the NMJ endplate. Neuromuscular blockers are clinically useful during surgery to facilitate tracheal intubation and provide complete muscle relaxation at lower anesthetic doses, allowing for faster anesthesia recovery and reducing postoperative respiratory depression.
(Curare), which native South American hunters of the Amazon region used to paralyze prey, was the first drug known to block the skeletal NMJ. (Tubocurarine), a drug which was developed as a result, but it has since been replaced by other agents with fewer side effects, such as (cisatracurium, pancuronium, rocuronium, and vecuronium).
A. Tubocurarine/ (prototype) is rarely used in clinical trials at the present time.
B. Metocurine (Metubine)/ is a tubocurarine derivative. It has the same properties, but with less histamine release and, as a result, less hypotension and bronchoconstriction. Metocurine's action lasts a long time (40 min). c. Atracurium (Tracrium) and Cisatracurium (Nimbex).
C. Atracurium/ Histamine is released in response to atracurium. It is inactivated in plasma spontaneously by nonenzymatic hydrolysis, which is slowed by acidosis. Hyperventilation-induced respiratory alkalosis shortens its duration of action. Laudanosine, a breakdown product of atracurium, can cause seizures if it accumulates.
D. Cisatracurium/ is an atracurium stereoisomer that produces less histamine and produces less laudanosine. In clinical practice, it has largely replaced the use of atracurium.
E. Mivacurium (Mivacron)/ is a short-acting (10–20 min) cholinesterase inhibitor that has a slower onset of action than succinylcholine and produces moderate histamine release at high doses.
F. Vecuronium (Norcuron), Rocuronium (Zemuron), and Pancuronium (Pavulon):
Because they are ineffective when taken orally, all neuromuscular-blocking agents are administered intravenously or intramuscularly. These agents have two or more quaternary amines in their bulky ring structure, which prevents them from being absorbed from the gut.
They have a poor ability to penetrate membranes and do not enter cells or cross the blood–brain barrier. Many drugs are not metabolized, and their effects are terminated by redistribution. Pancuronium, for example, is excreted unchanged in urine. Cisatracurium degrades naturally in plasma and through ester hydrolysis. [Note: Atracurium has been superseded by its isomer, cisatracurium.] Atracurium causes seizures by releasing histamine and being metabolized to laudanosine. Cisatracurium, which shares the same pharmacokinetic properties as atracurium, is less likely to cause these side effects]. Vecuronium and rocuronium, two amino steroid drugs, are deacetylated in the liver, and their clearance may be slowed in patients with hepatic disease. These drugs are also excreted in bile in their entirety. Drugs that are metabolized (e.g., mivacurium) or eliminated in bile (e.g., rocuronium) have shorter durations of action (10–20 min) than those eliminated by the kidney (e.g., metocurine, pancuronium, pipecuronium, and tubocurarine), which have durations of action of 35–60 min. The agent used is determined by the desired onset and duration of muscle relaxation.
Nondepolarizing agents are used as adjuncts to general anesthetics during surgery to cause muscle paralysis and relaxation. Not all muscles are equally sensitive to competitive agent blockade. The face and eye muscles are the most vulnerable and are paralyzed first, followed by the fingers, limbs, neck, and trunk muscles. The intercostal muscles are then affected, followed by the diaphragm. Muscles recover in the opposite direction. These agents are also used to control muscle contractions during electroconvulsive therapy and for muscle paralysis in patients when it is critical to control ventilation, such as ventilatory failure from pneumonia.
The Cardiovascular system: Because of histamine release, or ganglionic-blocking activity, tubocurarine, atracurium, mivacurium, pancuronium, and metocurine may cause cardiovascular effects such as hypotension or increased heart rate.
Respiratory system: Due to histamine release, some nondepolarizing agents can cause bronchospasm in sensitive individuals. Histamine-releasing agents are not recommended for asthmatic patients or those who have a history of anaphylactic reactions.
Acetylcholine and other depolarizing blocking agents work by depolarizing the plasma membrane of the muscle fiber. These agents, however, are more resistant to acetylcholinesterase (AChE) degradation and can thus depolarize muscle fibers for a longer period of time. Currently, the only depolarizing muscle relaxant in use is succinylcholine.
It is made up of two acetylcholine molecules that are linked end to end. Succinylcholine is metabolized in the liver and plasma by cholinesterase (butyrylcholinesterase or pseudocholinesterase). If given as a single dose, it has a few minutes of duration of action. Blockade may be prolonged in patients with plasma cholinesterase genetic variants that metabolize succinylcholine very slowly. These variant cholinesterases are resistant to dibucaine's inhibitory action. Acetylcholinesterase does not hydrolyze succinylcholine quickly.
Succinylcholine binds to the nicotinic receptor and depolarizes the junction in the same way that acetylcholine does. Unlike acetylcholine, which is instantly destroyed by AChE, the depolarizing agent remains at high concentrations in the synaptic cleft for a relatively long time, remaining attached to the receptor and providing constant stimulation of the receptor. [Note: Succinylcholine's duration of action is determined by diffusion from the motor endplate and hydrolysis by plasma pseudocholinesterase. Genetic variants that cause low or absent plasma pseudocholinesterase levels result in prolonged neuromuscular paralysis]. The depolarizing agent first opens the sodium channel associated with the nicotinic receptors, resulting in receptor depolarization (Phase I). This causes a brief twitch of the muscle (fasciculations).
Continued depolarizing agent binding renders the receptor incapable of transmitting further impulses. Continuous depolarization eventually gives way to gradual repolarization (Because tension cannot be maintained in skeletal muscle without periodic repolarization and depolarization of the endplate, continuous depolarization results in muscle relaxation and paralysis) when the sodium channel closes or becomes blocked This results in depolarization resistance (Phase II) and flaccid paralysis.
Administration of succinylcholine as an adjunct in surgical anesthesia to achieve muscle relaxation while using lower levels of general anesthetic, to induce brief paralysis in short surgical procedures, and to facilitate intubation.
Because of its rapid onset of action, succinylcholine is useful during the induction of anesthesia when rapid endotracheal intubation is required (a rapid action is essential if aspiration of gastric contents is to be avoided during intubation). It is also used during electroconvulsive therapy.
A. At higher doses, postoperative muscle pain may occur.
B. Hyperkalemia
Succinylcholine increases potassium release from intracellular stores, resulting in hyperkalemia from tissue potassium loss during depolarization.
Patients with burns, muscle trauma, massive tissue damage, or spinal cord transections are more likely to develop hyperkalemia because potassium is rapidly lost from within cells.
Hyperkalemia can be fatal, resulting in cardiac arrest and circulatory collapse.
C. Malignant hyperthermia
D. Apnea: Succinylcholine administration to a patient who is deficient in plasma cholinesterase or who has an atypical form of the enzyme can cause prolonged apnea (lasting 1–4 h) due to diaphragm paralysis in a small percentage of patients (1/10,000). In patients with electrolyte imbalances who receive this drug, the rapid release of potassium may also contribute to prolonged apnea. Succinylcholine should be used with caution or not at all in patients with electrolyte imbalances who are also receiving digoxin or diuretics (such as heart failure patients).
1. Whalen, K., Finkel, R. and Panavelil, T.A. (2015). Pharmacology. Philadelphia: Wolters Kluwer.
2. Tripathi, K.D. (2015). Essentials of medical pharmacology. New Delhi: Jaypee Brothers Medical Publishers (P) Ltd.
3. Katzung, B.G. (2018). Basic & clinical pharmacology. 14th ed. New York I 11 Pozostałych: Mcgraw-Hill Education, Copyright.
4. Rosenfeld, G.C. and Loose, D.S. (2014). Pharmacology. Baltimore, Md: Lippincott Williams & Wilkins.
5. Trevor, A.J., Katzung, B.G. and Marieke Kruidering-Hall (2015). Pharmacology examination & board review. New York: Mcgraw-Hill.
The term "cholinergic antagonist" refers to agents that bind to cholinergic receptors (muscarinic or nicotinic) and block the effects of acetylcholine
In order to understand Antimuscarinic drugs properly, it's required to classify them according to their chemistry.

The urethra is a muscular canal that extends from the neck of the bladder to the exterior of body. Read more about the anatomy of urethra in this article.

Dosage guide of Lisinopril: Click to read about the dose for your specific condition and age group.

Learn about medical uses, safety profile, mechanisms and interactions of statins.

Comprehensive guide on Ozempic (semaglutide), including its uses, dosage, side effects, warnings, and interactions.
.png)
Subscribe to our email lists and upgrade your life into a healthier and happier one.
SubscribeSign up for our newsletter(s) and receive healthy and scientific content to have a healthier life.
Choose Newsletter(s)Follow Medical Hex on social media platforms to be informed about latest health news and health-related scientific concepts.
A place you can find correct medical advice and information on health conditions as well as medical scientific articles. Our aim is to improve the health of human race by increasing health literacy through providing science-based content about health conditions.
©2022 Medical Hex. All Rights Reserved.
Choose what we can use. Strictly necessary and security cookies are always on.